Mulberry tree (Morus alba L.) is a species that belongs to the genus Morus and the family Moraceae. The genus holds around 16 species, with M. alba being the most globally distributed (Zhengyi & H. Raven, 2003), from temperate to subtropical regions of the Northern hemisphere to the tropics of the Southern hemisphere, including India and China as the two most important sites of cultivation (Ercisli & Orhan, 2007). Cultivation of M. alba is mainly used to feed silkworms, for which it is their main source of food, having a great importance in the silk industry. Extracts from their plant tissues have largely been used in traditional Chinese medicine due to their high bioactivity, attributed to the presence of phenolic compounds such as stilbenes. It was introduced to the Mediterranean basin in the 6th century, brought to Constantinople by monks, for silkworm-breeding. According to the legend its fruits were brought hidden because of the kept-secret lucrative production of silk. In Turkey, mulberries are also produced for human consumption of their raw fruit, as beverages or even syrup (Ercisli, 2004). On the Iberian Peninsula it has been cultivated as an ornamental and it has also been grown for the production of silk.
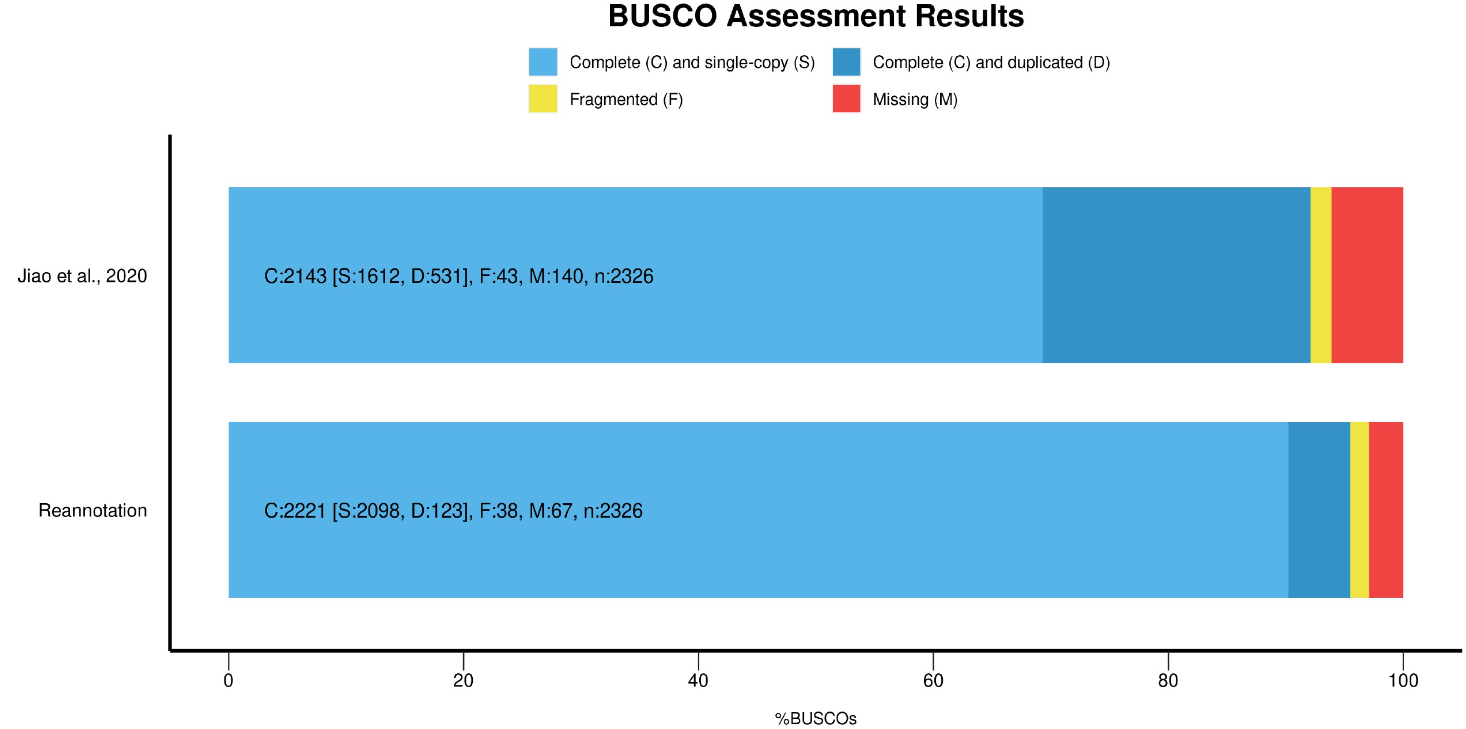
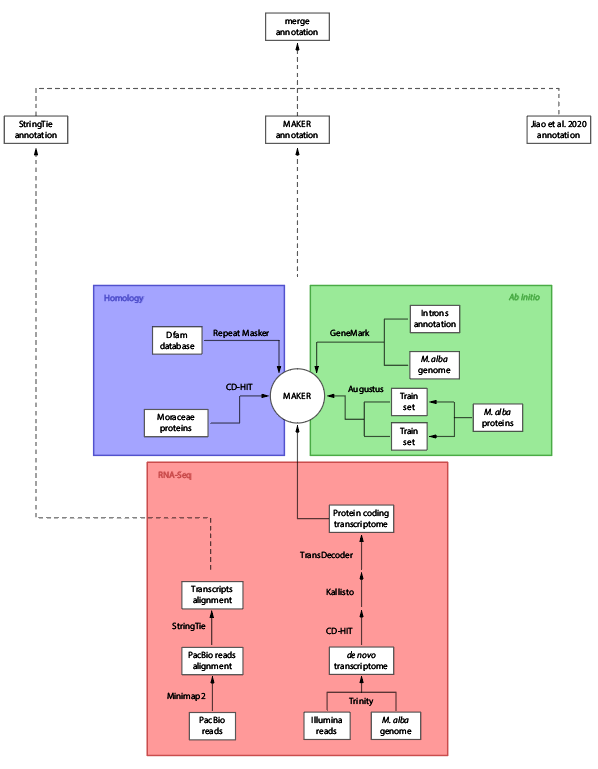
We followed an multi-step method to establish a V2.0 annotation of the mulberry genome; using a first version annotation by Jiao et al. (2020), we used a combination of 3 sources of evidence: Homology, ab initio prediction and RNA-seq. Firstly, the repetitive regions were masked using the Repeat Masker software. For the homologous prediction, the proteins from the Moraceae family in NCBI were downloaded to be mapped in the genome with BLASTx. The ab initio prediction was performed using Genemark and Augustus tools trained with some of the Jiao et al. (2020) annotated proteins. RNA-seq data coming from Illumina and PacBio sequencing was also used as evidence to predict genes. The Illumina reads were assembled in a de novo transcriptome for the aligment in the genome by tBLASTx. Minimap2 and StringTie were used to align the PacBio reads to the genome and recover the transcripts structure, respectivelly.
20,386 genes (Jiao et al. 2020)
31,401 genes (reannotation of the genome)